charmm_executable < charmm_script.inp > charmm_output.out
charmm –i 1.inp –o 1.out
RTF(residue topology file):包含了成键和电荷信息。Charmm包含了核酸,磷脂、蛋白和糖类的标准RTF文件
PARM(Parameter File):决定了能量:键长键角二面角力常数和范德华参数。Charmm包含了核酸,磷脂、蛋白、糖类和水的标准PARM文件
CRD(coordinates):体系的标准笛卡尔坐标
PSF(protein structure file):hold了成键和非键列表,键长键角二面角。对计算能量至关重要
标题必须有:
* This would be a short title
*
注释:
! this is a comment on a line by itself
结束:
Stop
蛋白,核酸,磷脂等的力场参数:
Charmm的力场参数要自行下载,放到输入文件所在文件夹下面供其调用https://www.charmm.org/charmm/showcase/news/new-protein-force-field/
小分子的力场参数:
1、通过amber的力场参数转化……………………blabla很难受!!但是,因为目前的任务是想把amber动力学之后的结构,用charmm来做计算,所以最好的方法是做力场转化。如果不转化的话,可能charmm的方法生成的小分子的原子类型之类的对不上,导致出错。(比如说加氢,amber是用的reduce,但是charmm我用的是gv保存的mol2)
2、charmm-gui(http://www.charmm-gui.org/?doc=input/ligandrm),提供了小分子的名字,可以自动生成其rtf文件和prm文件。PS:所用方法是CGenFF1.0版本。
3、直接用CgenFF(charmm小分子制作的标准方法,相当于antechamber)(https://cgenff.paramchem.org/initguess/#20181120_3/rg1.str)
(1)使用方法是先用gv保存分子的mol2文件,然后将mol2文件中的所有Ar替换为ar,在Molecule Name一行改为小分子的名字,然后导入分子,就可以直接生成。
(2)默认使用最新版本,目前的版本是2.2.0(使用的CgenFF版本是4.0),可以选择使用1.0版本(使用的CgenFF版本是3.0.1),输出结果与charmm-gui结果是一样的。
如:全部使用1.0版本:
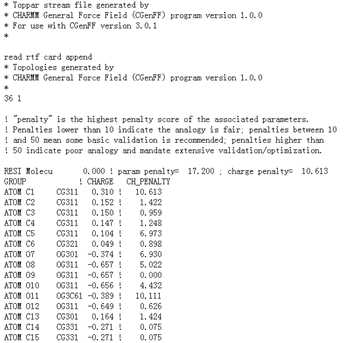
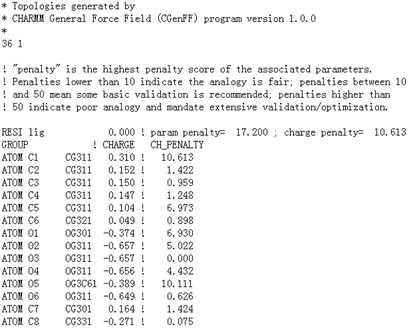
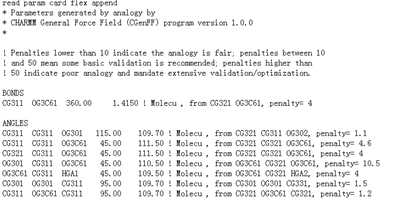
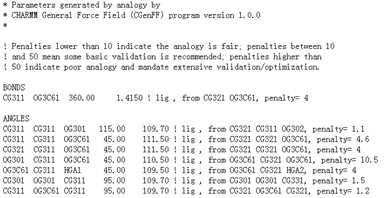
带有小分子的蛋白的动力学:
1、下载最新版本的蛋白质力场
2、下载与生成小分子相匹配的CGenFF力场,如:使用的CgenFF版本是2.2.0,则需要使用CgenFF的4.0版本。
3、下载水和离子的力场toppar_water_ions_namd.str
4、得到小分子力场my_ligands.str
5、读取参数文件和坐标
! read topology and parameter files
(1)With recent CHARMM force field releases, you can just read in the topology and parameter files as follows:
read rtf card name @TOPPAR/top_all36_prot.rtf
read param card flex name @TOPPAR/par_all36_prot.prm
read rtf card append name @TOPPAR/top_all36_cgenff.rtf
read para card flex append name @TOPPAR/par_all36_cgenff.prm
stream toppar_water_ions.str
(2)(optional) read in your own stream file:
stream my_ligands.str
! Read PSF and coordinates from file
(1)read psf card name all.psf
read coor card name all.crd
PS:可通过这个例子中的方法生成蛋白整体的psf和crd(https://www.charmmtutorial.org/index.php/Full_example)
原文:https://www.cnblogs.com/jszd/p/11178806.html