参考网址:http://software.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html
GSEA的图如何理解?
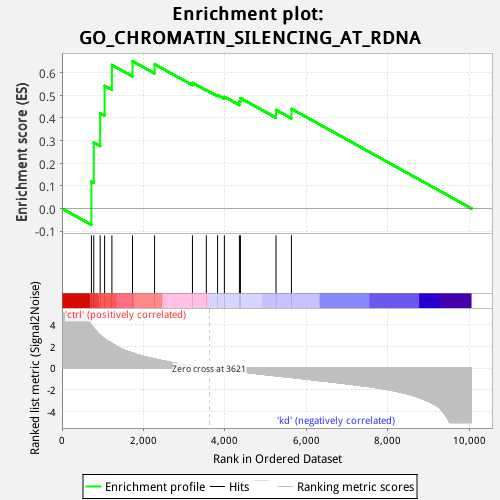
1. 图中下面灰色图的含义:http://software.broadinstitute.org/gsea/doc/GSEAUserGuideFrame.html?_Interpreting_GSEA_Results
图中的横坐标表示你输入的gene列表。
1)纵坐标含义:纵坐标表示“Ranked list metric”,GSEA官网对此的解释是ranking metric,即signal-to-noise ratio。
2)计算方法:
3)ranking metric的含义:它表示基因与表型(phenotype)的关系。即:这个基因与treat相关,还是与control相关,相关的度量值是多少。即ranking metric的含义。
positive raning metric表示与phenotype1相关,negative ranking metric表示与phenotype2相关。
表型是什么?比如,我们对给药组(treat)和正常组(control)做RNA-seq,得到每个基因在给药组和正常组中的表达值。在GSEA中,treat和control就是表型。
使用:
准备输入文件:
1. Expression dataset:是RNA-seq(或chipsiq)得到的数据。
通常有这么几列:
例1:NAME DESCRIPTION AML1 AML2 AML3 ALL1 ALL2 ALL3
TP53 na 681.3 638.0 665.0 240.0 587.0 737.0
例2:
NAME DESCRIPTION G C
TP53 na 681.3 638.0
注意:
1)必须用tab分隔;
2)第一行的名称必须是NAME和DESCRIPTION;
3)文件前两行必须是:
#1.2
53796(gene个数,即行数) 2(sample个数,不是phenotype数。如例1中phenotype数是2,sample数是6) 参考网址:http://software.broadinstitute.org/cancer/software/gsea/wiki/index.php/Data_formats
2. Phenotype labels文件:共三行。
2 2 1 分别为:sample数,phenotype数,1
# G C phenotype的名称
G C phenotype的名称
术语说明:
GSEA文档中的Phenotype、sample、gene sets等的含义。
Phenotype:
sample:
gene sets:
问题:
1.error:“Read timed out!”
这个错误,gsea官网没有说明。自己查的解决方法,费劲...。
原因:Gene sets database中,我选的是website的dataset,比如:ftp/...。这样,程序运行时,去GSEA的ftp服务器寻找并使用该文件。我怀疑可能网络不好,导致读取超时。
解决:下载gene set到本地。其实,就是把MSigDB的数据文件(.gmt文件)下载到本地。下载地址:http://software.broadinstitute.org/gsea/downloads.jsp。
注意:load本地的gene set文件时,只能通过“Load Data”功能执行。我在gene set database打开的界面找了好久都没找到load文件的图标,最后,灵机一动,才想到如何load。浪费时间%>_<%
2. error:After pruning, none of the gene sets passed size thresholds.
这个错误,gsea官网有说明。
原因:下载的c5.all.v7.0.symbols.gmt文件中的gene symbol全是大写,而我提供的expression文件中的基因名是小写。
解决:将expression文件的基因名改为大写即可。
具体解决方法:awk ‘{print toupper($3)}’,使用shell脚本的toupper函数即可。
3.运行时间:
我输入两个sample,两种phenotype,共53000+个gene的数据,permutation选择默认值1000,选择一个bp.gmtwenjian,运行时间长,大约十几分钟。
原文:https://www.cnblogs.com/zypiner/p/11777829.html