之前同组的同学已经在服务器上配置好了Intel Parallel Studio 2019,其中包括了Intel编译器以及mkl库,在本地的开发环境中并没有这些工具,这里使用Clion的远程调试功能。
在Tools->Deployment->Configuration中配置好SSH连接信息,同时选择通过SFTP进行文件传输。
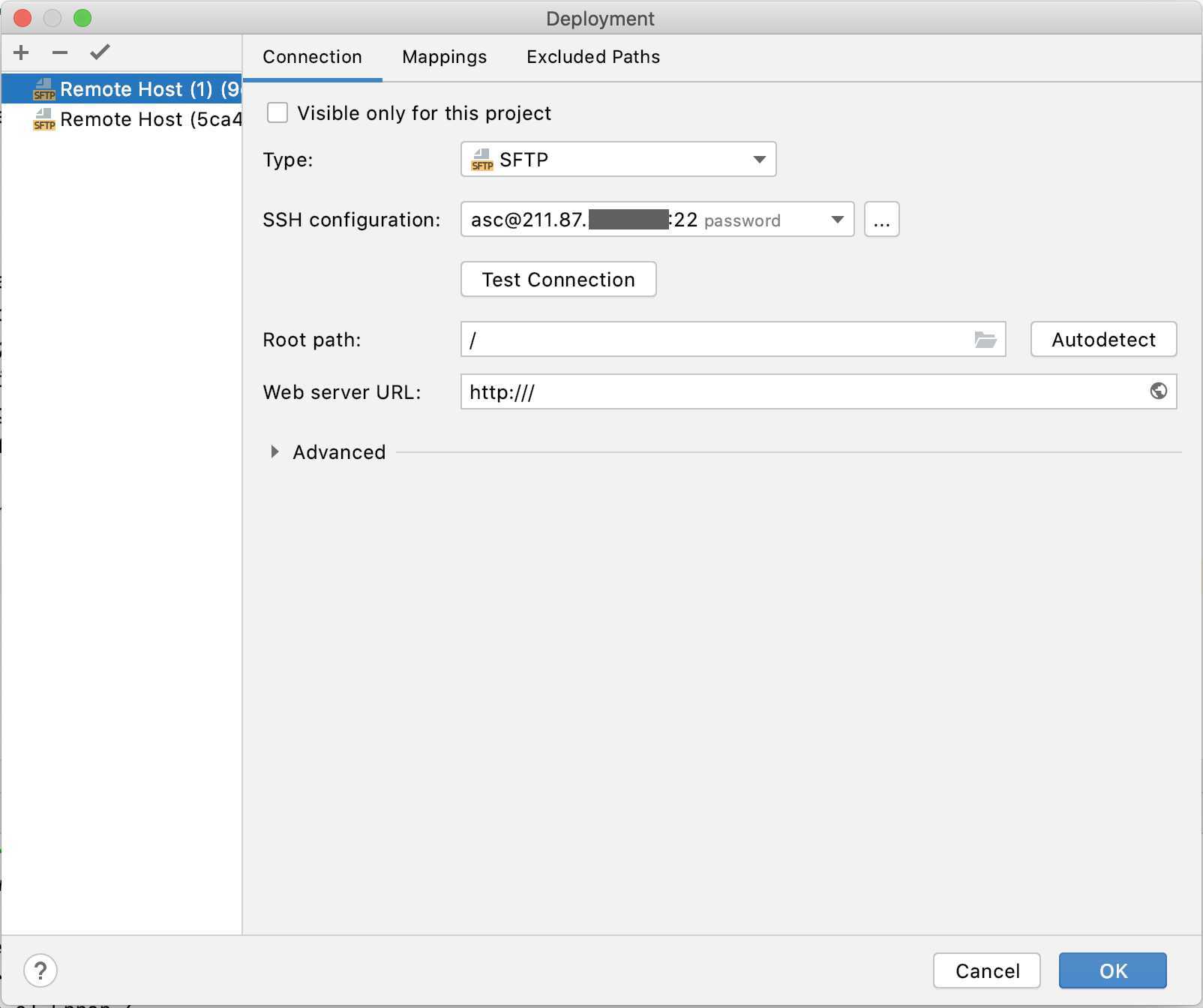
然后在Toolchains中配置好远程主机,并将其设为默认环境。
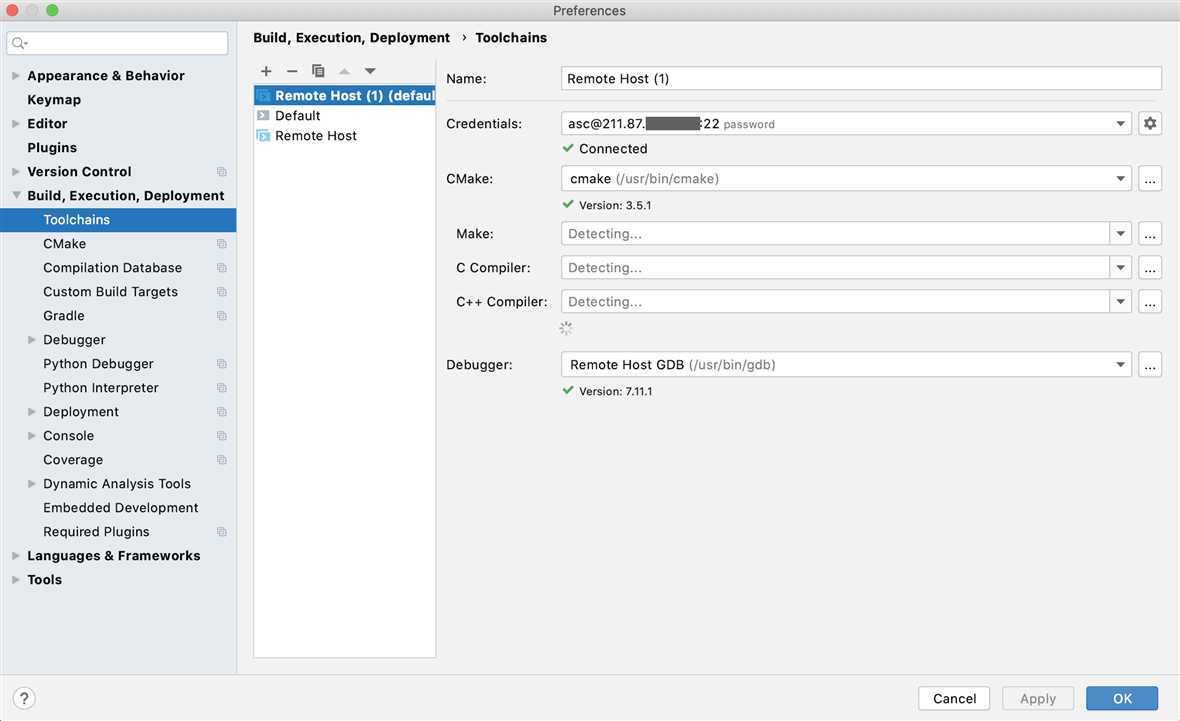
因为Clion并未添加Intel编译器支持,所以需要通过修改CMakeList.txt来使用Intel编译器,具体来说就是修改其中的CC和CXX,并且需要手动链接到mkl库所在目录。
#SET(CMAKE_CXX_COMPILER icpc)
SET(CMAKE_CXX_COMPILER /home/asc/intel/compilers_and_libraries/linux/bin/intel64/icpc)
#SET(CMAKE_C_COMPILER icc)
SET(CMAKE_C_COMPILER /home/asc/intel/compilers_and_libraries/linux/bin/intel64/icc)
link_directories(/home/asc/intel/mkl/lib/intel64 /home/asc/intel/lib/intel64)
配置完成后可以在本地和服务器进行文件同步,并且可以远程调试。
用户可以在控制台通过如下命令运行程序。
./newgwas -g `pwd`/../data/data10_0.bgen -s `pwd`/../data/ukb_imp_chr_v3.sample -o res.out -p height -c 10 --pca "pc1 pc2 pc3 pc4 pc5 pc6 pc7 pc8 pc9 pc10"
其中各个参数的意义如下:
| -g | 输入的bgen文件 |
|---|---|
| -o | 输出的结果文件 |
| -p | 选择分析的形状(必须出现在sample文件中) |
| -c | 每一轮的chunk大小 |
| --pca | pca参数(必须出现在sample文件中) |
通过cxxopts库解析用户输入的参数。

在程序运行的参数中,用户可以通过-o指定输出文件的位置:
在用户不指定输出位置时,默认输出到输入的bgen文件名+.result文件中:
if (result["output"].count()) {
ofilename = result["output"].as<string>();
} else {
ofilename = ifilename + ".result";
}
在main函数中,声明了std::vector<double> gwas_result;,计算完成后,将结果存储到gwas_result中,然后通过out_res函数输出程序执行结果。
bool out_res(std::string &ofilename,std::vector<NewGwas::Variant> &variants,std::vector<double> &res);
res中存放GLM求解得到的系数,后续可以添加更多结果。Variant是一个结构体,其中存放基因型相关数据。
struct Variant
{
std::string chromosome;
uint32_t position;
std::string rsid;
std::vector<std::string> alleles;
uint8_t * snps; //2-bit encode
};
首先处理并输出表头:
std::ofstream out(ofilename);
std::vector<string> title = {"rsid", "chromosome", "position", "alleleA", "alleleB", "index"};
for (int i = 0; i < res.size() / variants.size(); i++)title.emplace_back("coeff" + to_string(i));
for (auto &str:title)out << str << " ";
out << endl;
前部分输出形式与snptest相同。

然后每种基因型输出对应结果:
for (auto &it:variants) {
std::vector<string> data = {it.rsid, it.chromosome, to_string(it.position), it.alleles[0], it.alleles[1],to_string(++index),};
for (int i = (index - 1) * variants.size(); i < index * variants.size(); i++)
data.emplace_back(to_string(res[i]));
for (auto &str:data)out << str << " ";
out << endl;
}
输出完成后关闭文件流。

simple文件。
simple文件中存储的是表型数据,第一行是表型的名称,第二行是表型的种类。
第一列前两个ID分别对应样本的famliy ID和个体ID,第三列missing指的是样本的缺失率,第436列为协变量,第3745列为表型数据。
第二列代表变量的类型,前三项均为0,后面的字母对应含义如下:
| D | Discrete covariate (coded using positive integers) |
|---|---|
| C | 连续型协变量 |
| P | Continuous Phenotype |
| B | 二分类表型 |
这里使用R语言对simple文件进行可视化,首先读入文件
data<-read.csv("/Users/abc/Desktop/ukb_imp_chr_v3.sample",header = TRUE)
x_name<-names(data)
data<-data[[1]][-1]
其中,x_name存储列名,data存储具体数据。因为该文件不是标准csv格式,所以需要进行一些处理:
f<-function(x){
as.numeric(strsplit(gsub(‘NA‘, ‘0‘, x)[[1]], ‘ ‘)[[1]])
}
fx<-function(x){
x[[1]]
}
xx_name<-strsplit(x_name, ‘[.]‘)[[1]]
z<-lapply(data,f)
zx<-as.matrix(z)
znum<-apply(zx,1,fx)
znumx<-znum[,sample(ncol(znum),50,replace=F)]
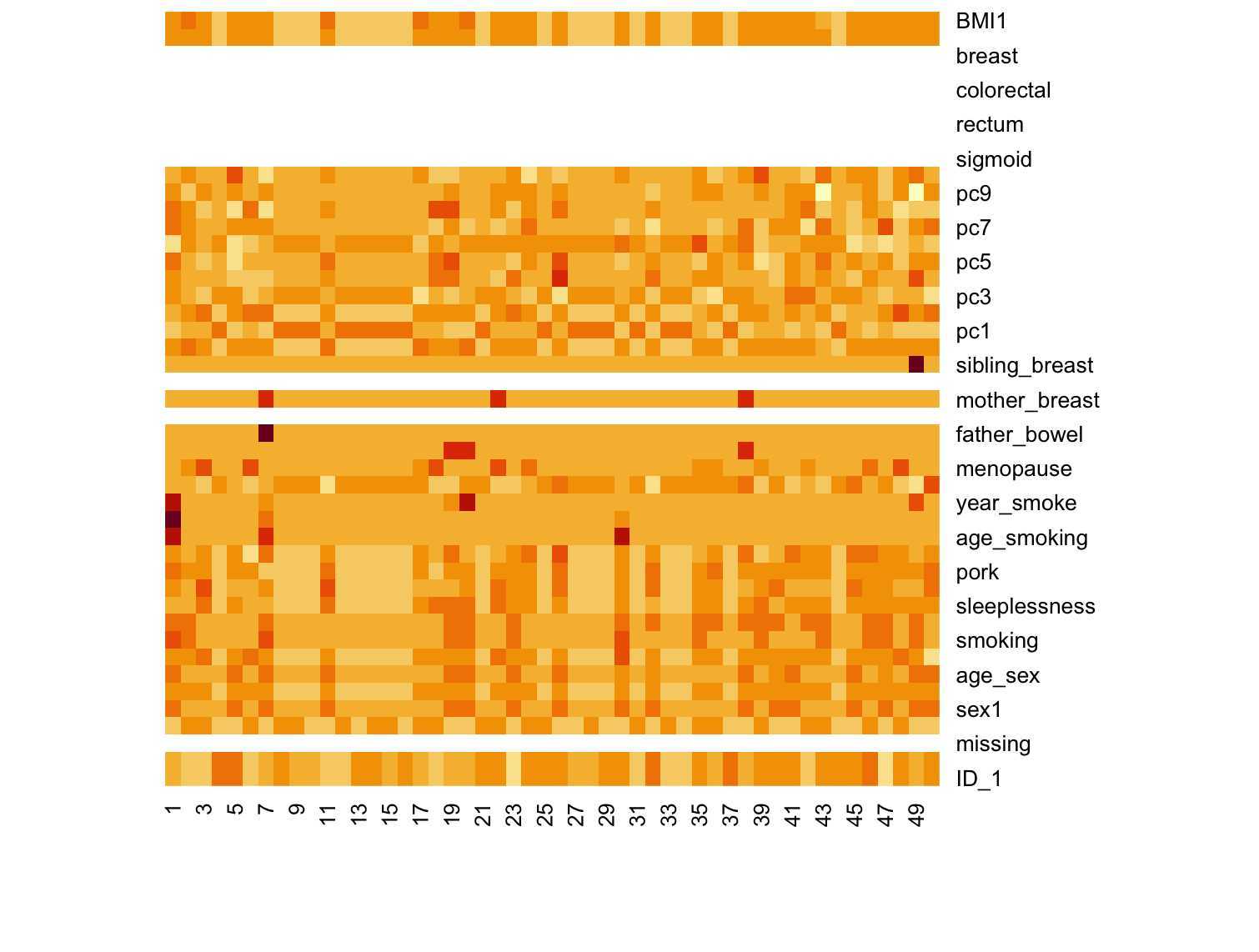
列名的中间是以.分隔开的,所以先用strsplit将其分隔成列表,f函数是将数据每行按照空格分隔开来,下面lapply方法将数据f函数应用到整个data列表。接下来将其转化成矩阵,然后将每一项单独提出来建立列表,最后从中抽取不重复的50项作为可视化的数据,然后通过heatmap函数生成可视化图片。
heatmap(znumx, Colv = NA, Rowv = NA,scale="row")
结果如下:
原文:https://www.cnblogs.com/DLAMDLAM/p/13222655.html